We use a number of analytical tools to perform metabolomic and lipidomic profiling. One of the powerful tools is the High-Performance Chemical Isotope Labeling (CIL) LC-MS Platform (the software for processing CIL LC-MS data is available from www.novamt.com). This platform is very sensitive and provides high metabolome coverage (thousands of metabolites). It is quantitative with high precision and accuracy, as isotope reagents are used for comparative analysis that overcome the matrix and ion suppression effects often associated with conventional LC-MS.
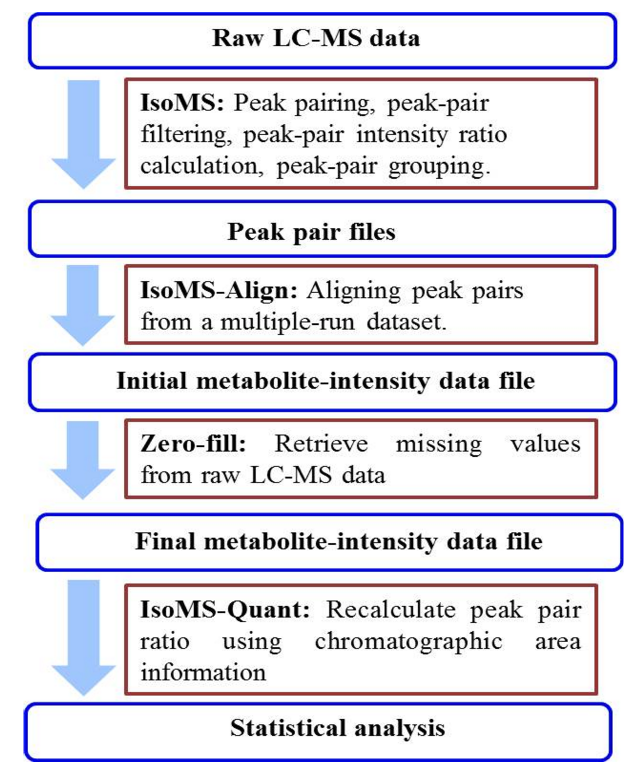
CIL LC-MS Data Analysis Workflow
The overall workflow for chemical isotope labeling (CIL) LC-MS metabolomics platform is shown below. Three programs, IsoMS, Zero-fill and IsoMS-Quant, are used in sequence to process the raw LC-MS data in batch mode to generate a metabolite-intensity data file that can be exported into a statistical tool for statistical analysis of the metabolomic profiles.